AlphaFold - The Most Useful Thing AI Has Ever Done
Based on Veritasium's video on YouTube. If you like this content, support the original creators by watching, liking and subscribing to their content.
Protein structure prediction became dramatically faster and more accurate when AlphaFold 2 combined evolutionary constraints with geometric reasoning rather than relying on brute-force folding or crystallography alone.
Briefing
AlphaFold turned protein folding—from a decades-long, expensive experimental grind into a near-automatic prediction task—by learning the rules of how amino-acid sequences become 3D structures. That shift matters because proteins are the molecular machines behind nearly every biological process, so knowing their shapes accelerates work in medicine, drug design, and even environmental engineering.
For more than six decades, researchers painstakingly determined the structures of roughly 150,000 proteins, largely through X-ray crystallography: crystallize a protein, shine X-rays, interpret diffraction patterns, and work backward to a structure. The bottleneck was brutal—protein crystallization can take years, and even a single structure can consume an entire PhD. The alternative, predicting structure from sequence, has long been a “holy grail” because proteins fold under a tangle of physical forces—electrostatics, hydrogen bonds, and solvent effects—yet evolution doesn’t design proteins from scratch.
A key obstacle was combinatorics. A short chain with 35 amino acids can fold into an astronomical number of configurations. Even checking tens of thousands of possibilities per nanosecond would take longer than the age of the universe to guarantee the correct structure. Competitions like CASP (launched in 1994) tried to force progress by rewarding models that output accurate structures without knowing the answers in advance. Early leaders such as Rosetta improved performance, and crowdsourcing efforts like Fold It even demonstrated that human intuition could solve specific protein puzzles—one HIV-related enzyme structure solved by more than 50,000 gamers.
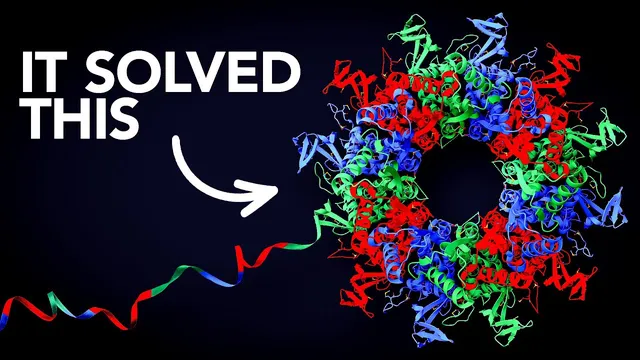
DeepMind’s breakthrough came when AlphaFold 2 replaced a simpler one-shot deep network with a more sophisticated architecture that fuses evolutionary clues and geometric constraints. AlphaFold 2 starts from the amino-acid sequence plus information derived from evolution: related proteins across species reveal which residues are conserved and which mutate together, a pattern called co-evolution. Instead of directly predicting a 3D structure, the system first learns a pairwise representation of residue-residue distances and orientations. Then the EVO Former—built around transformer-style attention—iteratively refines two “towers”: one for evolutionary relationships and one for geometry. Attention mechanisms let residues “talk” to each other, including triangular attention that enforces distance consistency through triangle-inequality constraints. A separate structure module then assembles the 3D arrangement by predicting how residue frames translate and rotate into place, with recycling loops that run the refinement multiple times.
In CASP 14, AlphaFold 2’s predictions for many proteins reached accuracy so high that they were virtually indistinguishable from experimentally determined structures, clearing the CASP threshold of 90. The payoff was enormous: within months, AlphaFold produced structures for nearly all proteins known to exist in nature—about 200 million—accelerating research across labs worldwide. Reported impacts include aiding malaria vaccine development, clarifying how mutations drive diseases ranging from schizophrenia to cancer, and helping study proteins in endangered species.
The story doesn’t stop at folding. The Nobel-winning protein design work associated with David Baker’s lab uses generative AI (including RF Diffusion) to create entirely new proteins for specific functions, such as designing human-compatible antibodies that can neutralize venom—potentially enabling scalable, transportable synthetic anti-venoms. More broadly, the transcript frames protein breakthroughs as a template for AI-driven science: once a hard “root” problem is cracked, entire branches of discovery can grow quickly—turning biology’s molecular complexity into something computationally tractable.
Cornell Notes
Protein folding is the long-standing problem of converting an amino-acid sequence into a protein’s 3D shape, which determines what the protein can do. AlphaFold 2 achieved near-experimental accuracy by combining evolutionary information (what residues are conserved or co-mutate across species) with geometric reasoning about residue distances and orientations. Its EVO Former uses transformer-style attention to iteratively refine evolutionary and structural predictions, including constraints like triangle inequality for distance consistency. A structure module then assembles the 3D protein using predicted translations and rotations, with multiple recycling passes to improve self-consistency. The result was CASP 14 performance that cleared the 90 threshold and enabled predictions for ~200 million proteins, accelerating medicine and protein engineering.
Why was predicting protein structure from sequence so hard before AlphaFold?
How did experimental methods like X-ray crystallography shape the field—and its bottlenecks?
What role did evolution-based data play in AlphaFold 2?
What changed architecturally from AlphaFold 1 to AlphaFold 2?
How did AlphaFold 2’s structure module generate 3D proteins?
What practical outcomes followed AlphaFold’s accuracy leap?
Review Questions
- How do evolutionary signals like conservation and co-evolution help constrain which residues must be near each other in a folded protein?
- Why does AlphaFold 2 benefit from transformer-style attention, and what does triangular attention enforce?
- What does the structure module predict (frames, translations, rotations), and why does recycling through the EVO Former matter?
Key Points
- 1
Protein structure prediction became dramatically faster and more accurate when AlphaFold 2 combined evolutionary constraints with geometric reasoning rather than relying on brute-force folding or crystallography alone.
- 2
X-ray crystallography remains expensive and slow because it depends on growing suitable protein crystals; this bottleneck limited how many structures could be solved for decades.
- 3
Levinthal’s calculation highlighted the combinatorial explosion of possible protein folds, making exhaustive search infeasible at realistic speeds.
- 4
AlphaFold 2’s EVO Former uses two interacting transformer-style “towers” (evolutionary information and geometry) and refines them through repeated information exchange cycles.
- 5
Co-evolution patterns across species help identify residue pairs that are likely close in 3D space, providing powerful training signals for folding.
- 6
AlphaFold 2’s structure module predicts residue frame translations and rotations, then recycles the result multiple times to improve self-consistency.
- 7
Protein design also advanced beyond folding: generative methods like RF Diffusion can create new proteins for functions such as producing human-compatible antibodies for neutralizing venom.